Dans un premier temps, nous allons comparer nos résultats à ceux obtenus
en 1991 par Landman et ses collaborateurs [52]. Ces derniers
avaient simulé la fission d'un agrégat
Na![]() , par une
dynamique moléculaire adiabatique pour les ions sur la surface de potentiel
Born-Oppenheimer, les électrons de valence de l'agrégat étant décrits
sur une grille tridimensionnelle de façon tout à fait similaire à ce
qui est présenté ici. Notre but est cependant d'étudier
une dynamique couplée des électrons et des ions, en supprimant
l'approximation adiabatique qui consiste à supposer
que les électrons de valence
sont à chaque instant dans l'état fondamental correspondant à la
géométrie ionique à cet instant.
, par une
dynamique moléculaire adiabatique pour les ions sur la surface de potentiel
Born-Oppenheimer, les électrons de valence de l'agrégat étant décrits
sur une grille tridimensionnelle de façon tout à fait similaire à ce
qui est présenté ici. Notre but est cependant d'étudier
une dynamique couplée des électrons et des ions, en supprimant
l'approximation adiabatique qui consiste à supposer
que les électrons de valence
sont à chaque instant dans l'état fondamental correspondant à la
géométrie ionique à cet instant.
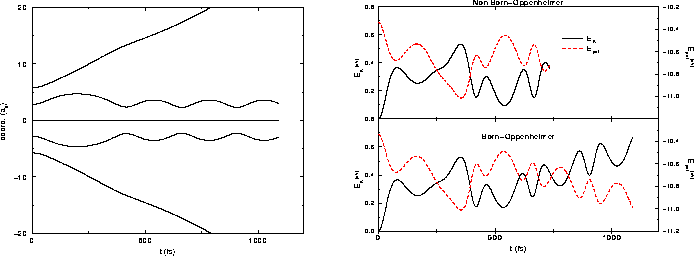
Nous présentons donc sur la figure III.9 un comparaison des
résultats obtenus avec l'approximation Born-Oppenheimer ou sans cette
approximation. La situation étudiée est très proche de celle de
[52]; la condition initiale auxiliaire est un agrégat
Na![]() , pris avec une géométrie de losange, subissant à l'istant
initial de la simulation une excitation modélisée par une double
ionisation qui enlève instantanément les fonctions
d'onde d'énergie les plus élevées : toute l'énergie d'excitation fournie
au système sert à l'ioniser. L'agrégat
Na
, pris avec une géométrie de losange, subissant à l'istant
initial de la simulation une excitation modélisée par une double
ionisation qui enlève instantanément les fonctions
d'onde d'énergie les plus élevées : toute l'énergie d'excitation fournie
au système sert à l'ioniser. L'agrégat
Na![]() résultant
est instable, (les
agrégats de sodium doublement chargés sont instables vis à vis de la
fission jusqu'à une taille de 27 atomes), et les résultats de dynamique
moléculaire présentés sur la figure III.9 ont été obtenus avec
cette condition initiale.
résultant
est instable, (les
agrégats de sodium doublement chargés sont instables vis à vis de la
fission jusqu'à une taille de 27 atomes), et les résultats de dynamique
moléculaire présentés sur la figure III.9 ont été obtenus avec
cette condition initiale.
|
On voit sur la figure III.9 que les dynamiques Born-Oppenheimer
comme non-Born-Oppenheimer donnent exactement le même résultat, c'est à
dire une explosion rapide en trois morceaux, un dimère
Na![]() qui reste seul dans la boîte et vibre transversalement,
et deux ions
Na
qui reste seul dans la boîte et vibre transversalement,
et deux ions
Na![]() qui sortent de la boîte de
calcul. Ces résultats sont tout à fait comparables aux résultats
originaux de [52].
qui sortent de la boîte de
calcul. Ces résultats sont tout à fait comparables aux résultats
originaux de [52].
Les deux méthodes donnent le même résultat car le nuage électronique se trouve à l'instant initial de la dynamique moléculaire dans son état fondamental. Les arguments de l'approximation Born-Oppenheimer sont valables ici et confirmés par le calcul non-Born-Oppenheimer : de pas de temps en pas de temps, les ions bougent car leur géométrie n'est pas stable, mais les fonctions d'onde électroniques suivent adiabatiquement ce changement de géométrie.
Ceci ne sera sûrement plus le cas si à l'instant initial le nuage électronique se trouve dans un état excité quelconque. Alors, il pourra y avoir échange d'énergie entre degrés de liberté ioniques et électroniques, avec conservation de l'énergie totale avec nos méthodes où la dynamique des électrons de valence est résolue en temps, ce qui ne serait pas vrai avec une simulation Born-Oppenheimer.