Le cas de
Na![]() est cependant très particulier,
pour les différentes raisons que nous avons exposées plus haut.
Il faut noter en outre qu'un consensus s'est établi sur la structure
ionique la plus probable pour cet agrégat ; comme de toute façon la
structure la plus stable dans le modèle du << jellium >> pour cet
agrégat est une sphère,
la plupart des structures ioniques possibles, à des différences
mineures près, rappellent cette sphère.
est cependant très particulier,
pour les différentes raisons que nous avons exposées plus haut.
Il faut noter en outre qu'un consensus s'est établi sur la structure
ionique la plus probable pour cet agrégat ; comme de toute façon la
structure la plus stable dans le modèle du << jellium >> pour cet
agrégat est une sphère,
la plupart des structures ioniques possibles, à des différences
mineures près, rappellent cette sphère.
Ces considérations ne sont plus valables pour d'autres agrégats, même
pour de petits agrégats de sodium.
Na![]() , entre autres, a
donné dans des expériences à très basse température des spectres de
photoabsorption extrêmement
fragmentés, et il ne semble pas pour le moment se dégager de consensus
entre théoriciens sur la structure la plus probable de cet agrégat.
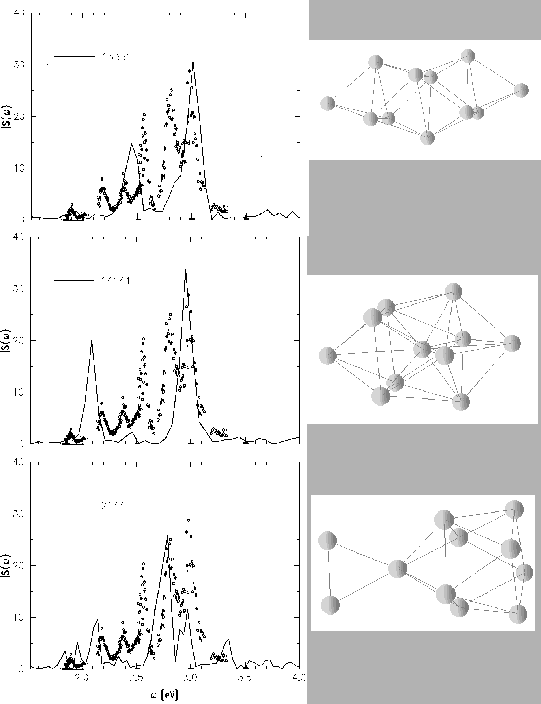
Le modèle CAPS, par exemple, a donné pour cet agrégat trois isomères
très proches en énergie, un état 14141 avec la symétrie
, entre autres, a
donné dans des expériences à très basse température des spectres de
photoabsorption extrêmement
fragmentés, et il ne semble pas pour le moment se dégager de consensus
entre théoriciens sur la structure la plus probable de cet agrégat.
Le modèle CAPS, par exemple, a donné pour cet agrégat trois isomères
très proches en énergie, un état 14141 avec la symétrie ![]() , une
configuration 13331 avec symétrie
, une
configuration 13331 avec symétrie ![]() et enfin une structure
2144 avec une très forte asymétrie droite-gauche. Ces trois structures
sont représentées sur la figure III.4, et la signification des
dénominations de la méthode CAPS y apparaît clairement : de gauche à
droite, on compte le nombre d'ions par plan à peu près orthogonal à l'axe de
symétrie, ici l'axe horizontal dans le plan de la figure.
Les pseudopotentiels employés ici n'ont pas la force des calculs CAPS
: nous avons pris une force différente, qui donne,
comme nous l'avons souligné dans le chapitre I, une position
du pic plasmon plus en accord avec les résultats expérimentaux ( ce
changement n'affecte pas la fragmentation des spectres). Les géométries
ont naturellement été réoptimisées pour ces nouveaux
pseudopotentiels, ce qui est revenu à une dilatation de quelques
% qui contribue également à amener les pics à une position en meilleur
accord avec les résultats expérimentaux.
et enfin une structure
2144 avec une très forte asymétrie droite-gauche. Ces trois structures
sont représentées sur la figure III.4, et la signification des
dénominations de la méthode CAPS y apparaît clairement : de gauche à
droite, on compte le nombre d'ions par plan à peu près orthogonal à l'axe de
symétrie, ici l'axe horizontal dans le plan de la figure.
Les pseudopotentiels employés ici n'ont pas la force des calculs CAPS
: nous avons pris une force différente, qui donne,
comme nous l'avons souligné dans le chapitre I, une position
du pic plasmon plus en accord avec les résultats expérimentaux ( ce
changement n'affecte pas la fragmentation des spectres). Les géométries
ont naturellement été réoptimisées pour ces nouveaux
pseudopotentiels, ce qui est revenu à une dilatation de quelques
% qui contribue également à amener les pics à une position en meilleur
accord avec les résultats expérimentaux.
|
On peut voir qu'aucun des isomères utilisés dans la figure III.4
ne donne de spectre comportant tous les pics visibles dans les résultats
expérimentaux. Contrairement au cas de
Na![]() , la restriction
à la symétrie axiale pour la structure ionique empêche d'avoir des
résultats convenables. On pourrait arguer que les contributions des
différents isomères présentés ont été observées simultanément dans les
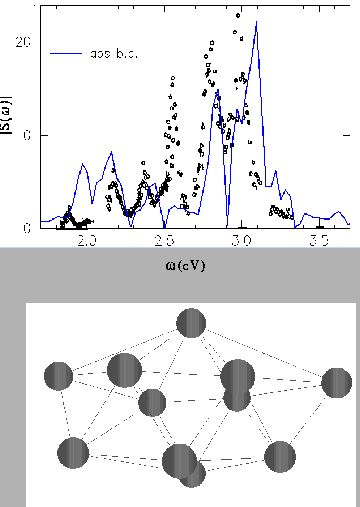
mesures expérimentales, mais sur la figure III.5, nous donnons le
spectre calculé avec un isomère, également représenté sur la figure,
obtenu par optimisation Monte-Carlo
tridimensionnelle par C.Kohl et P.G.Reinhard (communication privée).
, la restriction
à la symétrie axiale pour la structure ionique empêche d'avoir des
résultats convenables. On pourrait arguer que les contributions des
différents isomères présentés ont été observées simultanément dans les
mesures expérimentales, mais sur la figure III.5, nous donnons le
spectre calculé avec un isomère, également représenté sur la figure,
obtenu par optimisation Monte-Carlo
tridimensionnelle par C.Kohl et P.G.Reinhard (communication privée).
|
Pour ce calcul, effectué sur une grande grille, donc coûteux
numériquement, nous avons de plus utilisé les conditions aux limites
absorbantes qui sont décrites page ![[*]](cross_ref_motif.gif) . Puisque nous
sommes dans le régime linéaire, seule une faible partie de la densité
électronique est émise (moins de 5% du total), la position de la
résonance est donc à peine perturbée, mais l'usage de ces conditions
absorbantes permet d'augmenter le pas de temps utilisé dans la
simulation jusqu'à une valeur de 0.20 unités tout en assurant la
conservation de l'énergie, moyennant sa variation due à la faible
émission d'électrons. Ces conditions aux limites absorbantes augmentent
de plus légèrement la largeur des pics, par l'amortissement
supplémentaire du signal dipolaire qu'ils induisent.
. Puisque nous
sommes dans le régime linéaire, seule une faible partie de la densité
électronique est émise (moins de 5% du total), la position de la
résonance est donc à peine perturbée, mais l'usage de ces conditions
absorbantes permet d'augmenter le pas de temps utilisé dans la
simulation jusqu'à une valeur de 0.20 unités tout en assurant la
conservation de l'énergie, moyennant sa variation due à la faible
émission d'électrons. Ces conditions aux limites absorbantes augmentent
de plus légèrement la largeur des pics, par l'amortissement
supplémentaire du signal dipolaire qu'ils induisent.
Le résultat est meilleur que ceux obtenus avec structure ionique à symétrie axiale, mais n'est pas encore tout à fait satisfaisant. La position et le nombre des pics sont presque tous corrects, mais un effort d'optimisation des structures, et l'usage de pseudopotentiels encore meilleurs (sans doute non-locaux) semble nécessaire.
On peut conclure des résultats présentés ci-dessus qu'un calcul tridimensionnel complet est nécessaire en général pour calculer la structure et la réponse des agrégats de sodium ; notre méthode non-linéarisée, bien que motivée par l'étude de phénomènes dans les régime loin de l'équilibre, se révèle capable, moyennant suffisamment de soin dans le choix des pseudopotentiels et dans l'implantation numérique, de donner des spectres dans le régime linéaire largement aussi précis que ceux obtenus par des méthodes linéarisées, pour un coût comparable.